Pilot Study of Vaginal Microbiome Using QIIME 2™ in Women With Gynecologic Cancer Before and After Radiation Therapy
Objectives: To characterize the vaginal microbiome using QIIME 2™ (Quantitative Insights Into Microbial Ecology 2) in women with gynecologic cancer.
Sample & Setting: 19 women with gynecologic cancer before and after radiation therapy at a comprehensive cancer center in Atlanta, Georgia.
Methods & Variables: This pilot study analyzed vaginal microbiome communities using a microbiome analysis pipeline, beginning with 16S rRNA gene sequencing and processing through use of a bioinformatics pipeline to downstream microbial statistical analysis.
Results: The findings showed the methods to be robust, and most women with gynecologic cancer showed depletion of Lactobacillus. Compared to those pre–radiation therapy, women post–radiation therapy showed higher abundances of Mobiluncus, Atopobium, and Prevotella but lower abundances of Lactobacillus, Gardnerella, and Peptostreptococcus, which are associated with bacterial vaginosis.
Implications for Nursing: This study presents the fundamentals of human microbiome data collection and analysis methods to inform nursing science. Assessing the vaginal microbiome provides a potential pathway to develop interventions to ameliorate dysbiosis of the vaginal microbiome.
Jump to a section
The human body hosts trillions of microbiotas, including bacteria, viruses, fungi, and archaea; this number is close to that of human body cells (Knight & Buhler, 2015; Sender, Fuchs, & Milo, 2016). The human microbiotas and their genomes are collectively called the human microbiome (Ursell, Metcalf, Parfrey, & Knight, 2012), which varies among different hosts and across body sites within a single host (Morgan & Huttenhower, 2012; Spor, Koren, & Ley, 2011). Within the human microbiome, a number of potential biomarkers of cancer diagnosis, treatment, and prognosis have been identified (Rajagopala et al., 2017; Zitvogel, Ayyoub, Routy, & Kroemer, 2016), particularly regarding the roles of the human microbiome in treatment response and efficacy (Wilson & Nicholson, 2017), treatment-related toxicities, such as infections and pain (Kelly, Lyon, Yoon, & Horgas, 2016; Touchefeu et al., 2014), and disparities in treatment outcomes (Abbasi, 2017).
Nurses involved in clinical research and practice are greatly encouraged to have a general understanding of what the human microbiome is, how to measure the human microbiome, what microbiome findings are feasible, and what the applications of microbiome study findings are to clinical care (Claesson, Clooney, & O’Toole, 2017; Goodrich et al., 2014) because findings from current research may affect and be integrated with clinical care processes in the future. As microbiome science moves forward, nurse researchers and practitioners should have a general knowledge of computational analysis of microbiome data and ideally understand the process of human microbiome analysis. In the near future, clinicians may include assessment of a patient’s microbiome data in routine clinical practice. Several methods have been used to assess the human microbiome, such as the 16S rRNA gene, which includes nine variable regions (V1–V9) that provide a taxonomy profile of the microbiome (Ames, Ranucci, Moriyama, & Wallen, 2017). This study demonstrated methods for human microbiome analysis using 16S rRNA gene sequencing and QIIME 2™ (Quantitative Insights Into Microbial Ecology 2) bioinformatics pipeline and reported pilot results of the vaginal microbiome in women with gynecologic cancer before and after radiation therapy (RT).
Several packages for microbiome analysis are available, such as QIIME 2 (Navas-Molina et al., 2013), Mothur (Schloss et al., 2009), and RDP tools (Olsen et al., 1992). In this study, QIIME 2 is used as an exemplar tool for the vaginal microbiome analysis. QIIME 2 is a flexible microbiome analysis package to analyze 16S rRNA gene sequencing. Using the 16S rRNA gene sequence allows the comparison of the human microbiome communities to describe normal variations to pathologic disturbances (Caporaso et al., 2010).
Three aspects of the microbiome analysis that are important to examine consist of diversity (how many species are there [alpha diversity], and how similar are pairs of samples [beta diversity]?), taxonomy (who is there?), and abundance (how common or rare is a species relative to other species in a community?) (Claesson et al., 2017; Morgan & Huttenhower, 2012). QIIME 2 provides this information via different plugins. A plugin is an interface made available to QIIME 2 to support different functions of microbiome analysis. The original QIIME uses the operational taxonomic units (OTUs) being defined as clusters of reads that commonly differ by less than 3% (Hamady & Knight, 2009). QIIME 2 uses newly developed quality control methods (e.g., DADA2) to examine amplicon sequence variants (ASVs) to the single-nucleotide differences over gene sequencing regions (Callahan et al., 2016; Callahan, McMurdie, & Holmes, 2017). With the advancement of next-generation sequencing, new bioinformatics technologies, national initiatives of the Human Microbiome Project (HPM) and the American Gut Project (Human Microbiome Project Consortium, 2012; McDonald et al., 2018), studying the composition and functions of human microbiome and its translational applications is becoming an important component in clinical care and clinical research (McElroy, Chung, & Regan, 2017).
The healthy vaginal microbiome plays an important role in regulating microenvironmental disturbances and protecting against infections of the urogenital tract. A healthy woman’s vagina is primarily dominated by genus Lactobacillus (Blum, 2017). Lactobacillus species (spp.) help acidify the vagina to a pH of less than 4 by producing lactic acid, which is thought to provide protection against some sexually transmitted infections, including bacterial vaginosis (BV), human papillomavirus infection, and HIV/AIDS. A lower diversity is better for the vaginal microbiome; a dysbiotic vaginal microbiome is defined by increased vaginal microbiome diversity and the overgrowth of pathogenic microbial communities (Champer et al., 2018; Muls et al., 2017). A higher diversity of vaginal microbiome may interrupt the acidic vaginal environment. This is opposite from the gut microbiome, in which a higher diversity is better to maintain the gut microenvironmental hemostasis. Therefore, a low abundance of Lactobacillus spp. accompanied by polymicrobial anaerobic overgrowth, such as Gardnerella vaginalis, Prevotella spp., and Bacteroides spp., is associated with BV (Champer et al., 2018; Green, Zarek, & Catherino, 2015).
Cancer treatments, such as RT, in women with gynecologic cancer may result in a dysbiotic vaginal microbiome. Understanding the roles of the vaginal microbiome in cancer diagnosis, treatment, and outcomes is emerging and still in its nascent stage. Based on Bronfenbrenner’s ecological system theory (Bronfenbrenner, 1979), a multiple-factors framework has been proposed to study the human microbiome in cancer, including environmental factors (e.g., geographic locations, antibiotics exposure), family genetic factors, individual factors (e.g., race, lifestyles), and disease- and treatment-related factors (e.g., RT, cancer diagnosis) (Bai, Behera, & Bruner, 2017). This framework suggests that demographic factors (e.g., age, race) and treatment-related factors (e.g., RT) collectively contribute to a dysbiotic microbiome in cancer. A dysbiosis in the vaginal microbiome is associated with clinical outcomes in cancer, such as pain, sexual dysfunction, and urinary symptoms (Chase, Goulder, Zenhausern, Monk, & Herbst-Kralovetz, 2015). Until the results of the current study, characteristics of the vaginal microbiome pre- and post-RT were unknown in women with gynecologic cancer.
A healthy vaginal microbiome could promote health and decrease diseases. For women with gynecologic cancer, cancer treatments, such as chemotherapy and RT, may cause dysbiosis in the vaginal microbiome. Current understanding of this dysbiosis in women with gynecologic cancer is very limited. The advancement of next-generation sequencing (e.g., 16S rRNA) and bioinformatics tools (e.g., QIIME 2) provides means to study the diversity and compositions of the vaginal microbiome and its role in women’s experiences during cancer treatment. The purposes of this pilot study were to demonstrate the use of the QIIME 2 pipeline and provide a description of this technique with key concepts in the analysis process (see Figure 1). In addition, this study characterized the vaginal microbiome in a cohort of women with gynecologic cancer and explored the effect of demographic (e.g., age, race) and clinical (e.g., RT) variables on vaginal microbiome communities. 
Methods
This pilot study used a descriptive design to test methods for human microbiome analysis using 16S rRNA gene sequencing and microbiome identification via the QIIME 2 pipeline and to describe profiles of vaginal microbiome in a cohort of women with gynecologic cancer. This study was approved by the institutional review boards at Emory University and Grady Hospital in Atlanta, Georgia.
Sample and Setting
Eligibility criteria for women were being aged 18 years or older, being diagnosed with gynecologic cancer (i.e., endometrial, cervical, or vulvar cancer), being treated with RT with or without surgery and/or chemotherapy, and being able to speak English. Exclusion criteria included history of metastatic or other primary cancers other than gynecologic cancer and comorbidities that may cause severe vaginal toxicities (e.g., HIV/AIDS, autoimmune diseases, current sexual transmitted diseases, fungal infection). Use of antibiotics, oral or vaginal hormone replacement therapy, or corticosteroids within four weeks or spermicidal products within 48 hours of baseline assessment required rescheduling or exclusion. For medical pilot studies, a sample size of 10–30 is recommended for adequate ability to test hypotheses (Johanson & Brooks, 2010; Julious, 2005; van Belle, 2002). A convenience sample of 19 women with gynecologic cancer were enrolled in this study. All participants were recruited from Winship Cancer Institute, a comprehensive cancer center in Atlanta, Georgia.
Data Collection and Variables
Data collection occurred during patients’ clinical visits at the radiation oncology department. Trained research staff identified eligible patients from electronic health records and notified radiation oncologists one week before the potential patient’s clinic visit for RT consultation. The radiation oncologists introduced the study to eligible patients, including explaining the purpose of the study and its risks, and offered patients the opportunity to consider participation. Women interested in this study signed consent forms at their RT consultation. After the participants signed study consent forms, the clinical collaborators performed a pelvic examination and collected two vaginal swabs at the mid-portion of the patient’s vagina. Once collected, the swabs were immediately transferred to the attending research staff member who swirled and sealed the swabs in sterile Mo Bio PowerBead Tubes. The methods of vaginal microbiome data collection followed the Human Microbiome Project protocol (Human Microbiome Project Consortium, 2012). All vaginal microbiome samples were stored at –80°C at Emory University School of Nursing Biobehavioral Laboratory until DNA extraction.
The vaginal microbiome samples pre-RT were collected after diagnosis of malignancy and at least four weeks after surgery and pre-RT (T0). Follow-up samples were collected 1–2 months (T1), 2–4 months (T2), 4–6 months (T3), and 10–14 months (T4) post-RT. Patient demographic and clinical information were collected at T0.
DNA Extraction
The 16S rRNA gene sequencing is a primary method to identify the human microbiome in research and clinical settings (Ames et al., 2017). Raw DNA data extraction procedures followed the 16S gene preparation and sequencing protocol (Caporaso et al., 2011). DNA was extracted from vaginal swab samples stored in Mo Bio PowerBead Tubes using the Mo Bio PowerSoil® Isolation Kit to perform polymerase chain reaction amplification of 16S rRNA V3–V4 gene regions. For quality assurance, critical to human microbiome analysis, two biologic replicates from the first seven patients were generated at the Emory Integrated Genomics Core. Fifty-two 16S rRNA V3–V4 gene sequencing samples were sequenced. Because each V3–V4 gene sequencing sample has two sets of gene sequencing reads (one forward and one backward), the sequences were demultiplexed paired-end data (Renaud, Stenzel, Maricic, Wiebe, & Kelso, 2015) and resulted in a total of 104 sequencing reads for analysis.
QIIME 2 Pipeline
The QIIME 2 pipeline generates the bacterial communities’ information for each sample. The process includes two stages, referred to as the upstream and downstream phases (see Figure 2). The upstream stage consists of importing 16S rRNA sequences, ensuring sequences quality control, constructing the feature table, and generating the phylogenetic tree. In the feature table, each value indicates the frequency of a feature for the corresponding sample. The downstream stage consists of taxonomic, diversity, and abundance analysis (Caporaso et al., 2010). In this phase, statistics and interactive visualizations of the data are used (Navas-Molina et al., 2013). 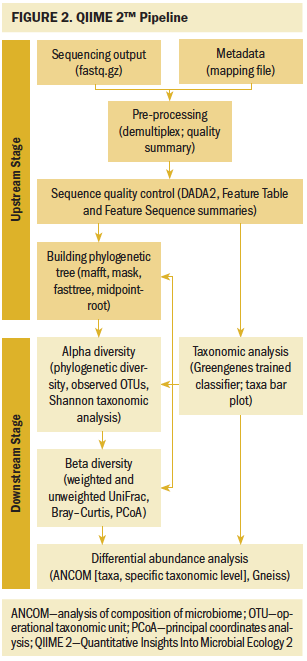
Upstream stage processing: The upstream stage started with importing 16S rRNA gene sequences. The V3–V4 gene sequences were checked for appropriate names and data format following QIIME 2 requirements. To do this, 16S rRNA gene sequences and a metadata file with key clinical variables were imported into QIIME 2. The metadata file provided the descriptive data (e.g., race, RT) associated with gene sequences. The 16S rRNA reads were imported into QIIME 2 for analysis.
The next step was sequence quality control. QIIME 2 provides several quality control methods, including DADA2 (Callahan et al., 2016), Deblur (Amir et al., 2017), and Phred quality score–based filtering to remove or correct sequencing reads with errors or chimeras. A higher Phred quality score indicates better nucleotide quality for gene sequences (Blankenberg et al., 2010). The Phred quality score ranges from 0–42. A quality score higher than 20 means less than 1% probability error. In the current study, DADA2 was chosen to correct amplicon sequence errors and filter the chimeric sequences. The chimeric sequences reflect sequencing artifacts rather than real biologic diversity.
The third step, building the phylogenetic tree, shows the relationships of different bacterial species to each other in a tree-like fashion that includes nodes, implying a common descendant, and branches, implying a split from the descendant. Bacterial species that are more similar will be closer in proximity to each other on the tree. The phylogenetic tree is essential for microbial diversity analysis.
Downstream stage processing: The first step in the downstream stage is taxonomic analysis. This analysis matches the DNA sequence to a microbial taxon from phylum to species levels. QIIME 2 comes bundled with Greengenes reference database. The authors implemented a trained classifier tailored to the gene region using V3–V4 Greengenes reference database and classified the taxa of the representative sequences (Werner et al., 2012). Trained classifier is a trained marker gene reference database that is used for taxa classification.
Next, diversity analysis was performed. Diversity analysis can assess within-sample diversity (alpha diversity) and between-sample diversity (beta diversity). QIIME 2 can create several alpha diversity metrics: observed OTUs (bacterial community richness), Shannon’s index (bacterial community richness and evenness), Faith’s phylogenetic diversity (PD) (bacterial community richness that incorporates phylogenetic relationships between taxa), and Pielou’s species evenness (bacterial community evenness) (Lozupone & Knight, 2005). Richness means the number of species present in a sample and evenness represents the relative abundance of different species that make up the richness. Beta diversity operates on a pair of samples (Caporaso et al., 2012; Lozupone & Knight, 2005). Common beta diversity metrics include Bray–Curtis distance (abundance without phylogeny), Jaccard distance (presence and absence of OTUs without phylogeny), unweighted UniFrac distance (presence and absence of OTUs with phylogeny), and weighted UniFrac distance (abundance of OTUs with phylogeny).
The final process of the downstream stage is abundance analysis. The abundance analysis discriminates differentially abundant taxa based on variables of interest, such as race and RT. QIIME 2 provides multiple mechanisms (e.g., analysis of composition of microbiome [ANCOM]) (Mandal et al., 2015) for this analysis. According to ANCOM requirements, the authors filtered out the taxa that only appear in one sample and taxa counts less than 10 across all samples.
Data Analysis
Bacterial taxonomies were assigned using the pretrained 16S rRNA V3–V4 classifier based on the Greengenes reference database. Alpha and beta diversity metrics were used to describe the composition of the vaginal microbiome. Comparisons of alpha diversity were conducted using Kruskal–Wallis test for categorical data (race and RT) and Spearman’s rank correlation coefficient for continuous data (age and total RT dose). Permutational multivariate analysis of variance (PERMANOVA) (Tang, Chen, & Alekseyenko, 2016) was used to test the associations between microbial beta diversity and demographic and clinical variables (race and RT). Principal coordinates analysis (PCoA) was used to visualize sample dissimilarities (Caporaso et al., 2010; Vázquez-Baeza, Pirrung, Gonzalez, & Knight, 2013) based on the Bray–Curtis and unweighted UniFrac distance metrics. ANCOM was used to examine differentially abundant taxa in terms of race and RT. All data analyses were completed using QIIME 2. The full dataset and analyzing results can be found on GitHub (https://bit.ly/2MtqCr0).
Results
Participant Characteristics
Fifty-two vaginal swab samples from 19 women with gynecologic cancer were analyzed. These vaginal microbiome samples were collected at T0 (n = 17) or T1–T4 (n = 35). The median age was 62 years (range = 36–71 years). Participants were African American (n = 14), Caucasian (n = 3), Latina (n = 1), and Asian (n = 1). The women had been diagnosed with endometrial (n = 10), cervical (n = 8), or vulvar cancer (n = 1). Five women received external beam RT, seven women received high-dose rate brachytherapy, and seven patients received a combination of both treatments. Mean total dose of RT was 54.21 Gy.
Upstream Stage Processing
Among 52 vaginal swab samples, the sequence count per sample ranged from 85,872 to 892,422, with a mean sequence count of 412,014 per sample. Based on the Phred quality score greater than 20, the raw sequences were trimmed at 17 and 21 base pairs (for the length of primers) and truncated at 250 and 250 base pairs (for poor quality scores) for forward and reverse 16S rRNA V3–V4 sequencing reads, respectively. After the DADA2 process, 19,365 features were reported. Frequencies per feature ranged from 1 to 735,173; feature frequencies per sample ranged from 5,225 to 192,353.
Downstream Stage Processing
Taxonomic analysis: QIIME 2 visualizes taxonomic findings using the taxa-bar-plot, whose stacked bar graph output joins sample metadata to summarize the relative frequency (%) of taxa present specifically at each taxon level, such as phylum and genus. Figure 3 describes the microbial taxonomy at the phylum level. In this study, only 18 of 52 samples had greater than 10% dominance of Lactobacillus. 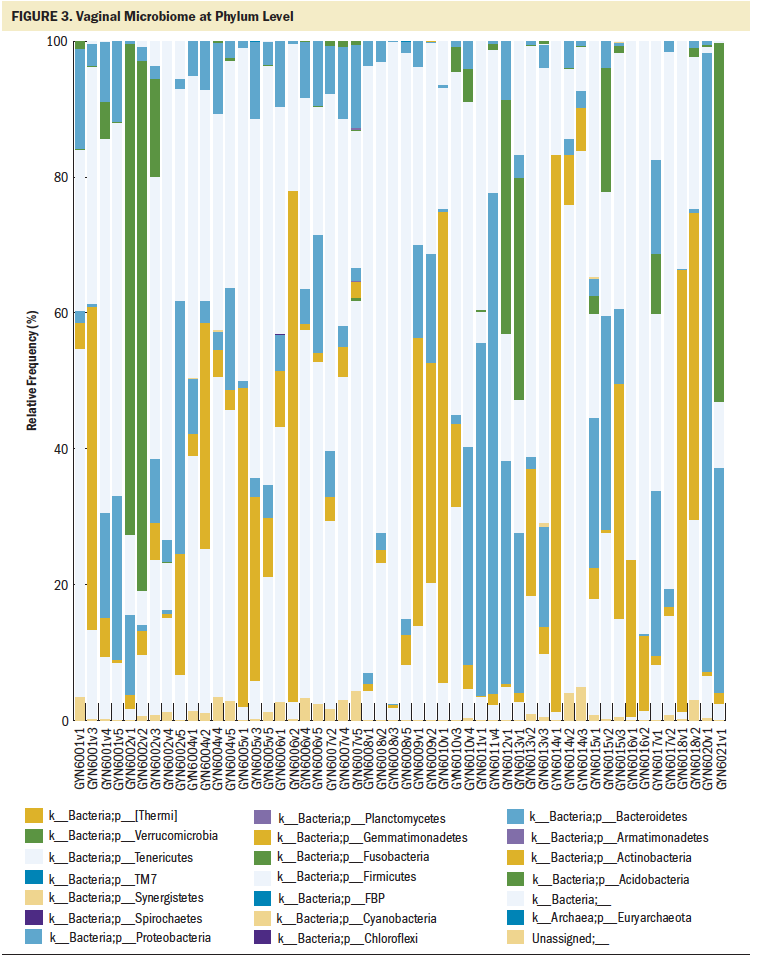
Diversity analysis: Based on Spearman’s rank correlation coefficient, age and total dose of RT were not associated with any of the alpha diversity metrics; RT (pre and post) and race (Caucasian, African American, Asian, and Latina) were associated with alpha diversity metrics. Compared to those pre-RT, women with gynecologic cancer post-RT showed a higher alpha diversity in Shannon’s index (p = 0.01), observed OTUs (p = 0.001), Faith’s PD (p = 0.001), and Pielou’s species evenness (p = 0.04). Race was significantly associated with alpha diversity metrics (Shannon’s index: p = 0.02; observed OTUs: p = 0.02; Faith’s PD: p = 0.02; Pielou’s species evenness: p = 0.03) based on the Kruskal–Wallis test. Caucasian women with gynecologic cancer showed higher trends in alpha diversity than African American women (Shannon’s index: p = 0.06; observed OTUs: p = 0.07; Faith’s PD: p = 0.07).
PERMANOVA showed significant associations between beta diversity metrics and race and RT. Significant differences of vaginal microbiome metrics were found between pre- and post-RT in Bray–Curtis distance (p = 0.02), Jaccard distance (p = 0.04), and unweighted (p = 0.002) and weighted (p = 0.01) UniFrac distances. Race was significantly associated with the vaginal microbiome metrics in Bray–Curtis distance (p = 0.003), Jaccard distance (p = 0.003), and unweighted (p = 0.008) and weighted (p = 0.05) UniFrac distances. Based on Bray–Curtis distance and unweighted UniFrac distance matrices, PCoA showed dissimilarities of vaginal microbiome with respect to RT (pre and post) and race (African American, Caucasian, Asian, and Latina).
Abundance analysis: Among microbial taxa associated with BV (Onderdonk, Delaney, & Fichorova, 2016), ANCOM showed that women post-RT had higher abundances of the taxa Mobiluncus, Atopobium, and Prevotella, but lower abundances of Lactobacillus, Gardnerella, and Peptostreptococcus than pre-RT (see Table 1). The only differential taxa abundance related to race was Brucellaceae at the family level. 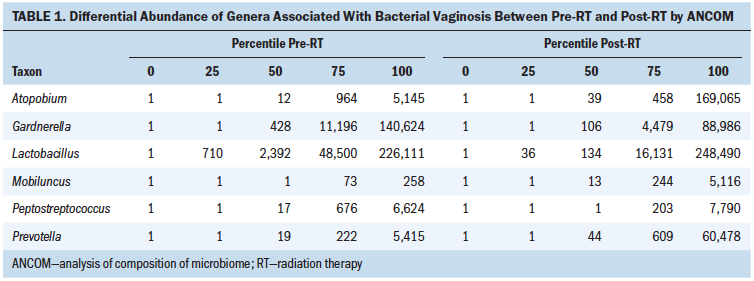
Discussion
This study demonstrates the fundamental methods for vaginal microbiome analysis using QIIME 2. The findings indicated that RT and race were associated with diversity of the vaginal microbiome. Women post-RT showed higher abundances of pathogenic microbes associated with BV. This pilot work provides first-hand evidence regarding the effect of RT on the vaginal microbiome and supports the need for ongoing study to further understand the determinants and consequences of dysbiotic vaginal microbiome in women with gynecologic cancer.
Using 16S rRNA and QIIME 2 to analyze vaginal microbiome data in women with gynecologic cancer is feasible based on standard protocols (Human Microbiome Project Consortium, 2012). The study team found QIIME 2 useful for microbiome analysis. The team installed it on several computational platforms, such as a native Linux server, Amazon Web Services, VirtualBox on Windows, and MacOS operating system. As a no-cost bioinformatics pipeline, QIIME 2 has multiple strengths, including straightforward installation process, automatic assignment of provenance (capturing actions and details associated with each step of data analysis) to increase the rigor and reproducibility of microbiome data analysis, sharing findings with and without QIIME 2 installation, and online data visualization. Therefore, QIIME 2 can be easily adapted to analyze any type of microbiome data in nursing science.
Quality assurance is essential for human microbiome studies (Goodrich et al., 2014). Variations from microbiome sample collection, storage, and analysis can lead to bias in study findings (Sinha, Abnet, White, Knight, & Huttenhower, 2015; Sinha et al., 2017). In this model of vaginal microbiome analysis using QIIME 2, several actions were taken to ensure the quality of the analysis. Team members of the microbiome analysis group have attended a QIIME 2 workshop hosted by the QIIME 2 team. In addition, all the steps using QIIME 2 in this study were tested based on a QIIME 2 tutorial, with support of the QIIME 2 development team. The team members have independently analyzed the same vaginal microbiome data attached with the metadata using the same QIIME 2 and trained classifier. Comparisons of final results among group members showed identical results, supporting the validity and robustness of QIIME 2 for the vaginal microbiome analysis.
Based on the multiple-factors framework (Bai et al., 2017; Chase et al., 2015), a variety of factors, such as demographics and cancer treatment, can significantly contribute to dysbiosis of the vaginal microbiome. RT has long been used as a curative or palliative therapy in gynecologic cancer and can potentially disturb compositions of the vaginal microbiome (Muls et al., 2017). This pilot study showed depletion of Lactobacillus pre- and post-RT. Lactobacillus is thought to be the dominant genus in the vaginal microbiome of premenopausal (Gajer et al., 2012; Ravel et al., 2011) and postmenopausal women (Hummelen et al., 2011). Anaerobic metabolism of glycogen by vaginal epithelial cells and vaginal microbes, such as Lactobacillus, produce low vaginal pH. The resulting acidic environment can protect against pathogenic microbes of the genitourinary tract (Boskey, Cone, Whaley, & Moench, 2001). In the absence of Lactobacillus, pathogenic microbes associated with BV were often predominant (Fredricks, 2011). As is seen in women with symptomatic BV, when Lactobacillus was at low abundance, a diverse group of pathogenic bacteria were seen in its place (Muhleisen & Herbst-Kralovetz, 2016; Onderdonk et al., 2016). The current findings in women with gynecologic cancer seem to show vaginal microbiome compositions consistent with the literature among women with symptomatic BV (Muhleisen & Herbst-Kralovetz, 2016; Onderdonk et al., 2016), showing low abundance of Lactobacillus but higher abundances of BV-associated pathogens, such as Atopobium and Prevotella. These findings suggest a higher risk of BV among women with gynecologic cancer post-RT.
This study showed an increase in vaginal microbiome diversity from pre- to post-RT; as reported previously, the increased diversity was associated with pathogenic microbes, such as Atopobium and Prevotella. From this small sample size, the findings showed differences in the vaginal microbiome diversity related to race, which aligned with previous findings in healthy women (Fettweis et al., 2014). Further research is needed to understand the potential role of the vaginal microbiome in treatment outcomes, particularly in the poorer outcomes documented in African American women treated for gynecologic cancer (Cote, Ruterbusch, Olson, Lu, & Ali-Fehmi, 2015). Collectively, the findings seemed to support the effect of RT on the vaginal microbiome, which may potentially lead to poor treatment-related outcomes.
Limitations
This study suggested that women with gynecologic cancer have, relative to reports in the literature (Hummelen et al., 2011), abnormal vaginal microbiome profiles pre- and post-RT. These findings should be further tested in a large and diverse sample of women with gynecologic cancer compared to a similarly diverse group of healthy women. A healthy control group can help mitigate the effect of menopausal status and other confounders, such as race, sexual behaviors, use of antibiotics, prebiotics, probiotics, and intravaginal products. In addition, as one of the first studies to report the vaginal microbiome in patients with gynecologic cancer pre- and post-RT, the current analysis focused on genus level of Lactobacillus rather than species level, which may preclude further interpretation of the relationships between specific species of the vaginal microbiome (e.g., Lactobacillus crispatus, Lactobacillus jensenii, Lactobacillus iners) and treatment-related outcomes, such as BV. Despite these limitations, this study introduced new knowledge regarding human microbiome data collection and analysis, which will help inform clinical nurses about future directions in cancer care.
Implications for Nursing
This study reported the methods for vaginal microbiome analysis, including 16S rRNA sequencing and the QIIME 2 pipeline. The methods presented in the current article can be generalized to study other microbiome data of interest, such as gut, oral, and skin microbiomes. As a new field in cancer, this study provides a promising opportunity for nursing scientists to understand the biologic characteristics of the vaginal microbiome on cancer diagnosis, treatment, and prevention. Identification of specific microbial communities may lead to precise therapies, such as prebiotics and probiotics, which may play a role in preventing gynecologic cancer or ameliorating gynecologic cancer treatment–related toxicities (e.g., vaginitis, vaginal fibrosis, sexual dysfunction) (Bahng, Dagan, Bruner, & Lin, 2012; Bruner et al., 1993).
Microbiome-related work has moved into clinical settings. 16S rRNA sequencing and microbiome analysis have been used to identify resistant microbial pathogens, reducing the prevalence of hospital-acquired infections (Ames et al., 2017).
The vaginal microbiome seems to play crucial roles in high-grade cervical dysplasia and cervical carcinoma (Kyrgiou, Mitra, & Moscicki, 2017; Piyathilake et al., 2016). Knowledge regarding the vaginal microbiome communities can help detect and monitor gynecologic cancer occurrence and progression. The complex role of the human microbiome in cancer care is beginning to be understood. Direct clinical applications of the human microbiome are still limited. Understanding the promising clinical implications of the human microbiome and bioinformatics techniques associated with the human microbiome are of particular relevance to nurse scientists and clinical practitioners, who are uniquely positioned to study and use these techniques in clinical settings. 
Conclusion
This study introduced the use of 16S rRNA sequencing and QIIME 2 to analyze vaginal microbiome data among women with gynecologic cancer and suggested that QIIME 2 is a robust and valid tool to inform nurse scientists in human microbiome analysis. Pilot findings showed that RT may lead to a higher diversity of vaginal microbiome and a higher abundance of pathogenic microbiotas associated with BV. Future studies are needed to understand the effect of the vaginal microbiome on cancer treatment–related toxicities and outcomes and to develop precise interventions to promote a healthy microbiome.
About the Author(s)
Jinbing Bai, PhD, MSN, RN, is an assistant professor in the Nell Hodgson Woodruff School of Nursing; Ileen Jhaney, MSPH, is a research informatics analyst in the Winship Cancer Institute; and Gaea Daniel, MSN, RN, is a PhD candidate and Deborah Watkins Bruner, PhD, RN, FAAN, is a professor, both in the Nell Hodgson Woodruff School of Nursing, all at Emory University in Atlanta, GA. This research was funded, in part, by the Emory Integrated Genomics Core Shared Resource of Winship Cancer Institute of Emory University and the National Institutes of Health/National Cancer Institute (No. P30CA138292) and by a Friends of Winship Fashion Research Scholar Award from the Winship Cancer Institute (principal investigator: D.W. Bruner). Bai, Daniel, and Bruner completed the data collection and provided statistical support. All authors contributed to the conceptualization and design, provided the analysis, and contributed to the manuscript preparation. Bai can be reached at jinbing.bai@emory.edu, with copy to ONFEditor@ons.org. (Submitted February 2018. Accepted October 1, 2018.)
References
Abbasi, J. (2017). For health disparities, don’t ignore the microbiome. JAMA, 317, 797. https://doi.org/10.1001/jama.2017.0447
Ames, N.J., Ranucci, A., Moriyama, B., & Wallen, G.R. (2017). The human microbiome and understanding the 16S rRNA gene in translational nursing science. Nursing Research, 66, 184–197. https://doi.org/10.1097/NNR.0000000000000212
Amir, A., McDonald, D., Navas-Molina, J.A., Kopylova, E., Morton, J.T., Zech Xu, Z., . . . Knight, R. (2017). Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems, 2(2). https://doi.org/10.1128/mSystems.00191-16
Bahng, A.Y., Dagan, A., Bruner, D.W., & Lin, L.L. (2012). Determination of prognostic factors for vaginal mucosal toxicity associated with intravaginal high-dose rate brachytherapy in patients with endometrial cancer. International Journal of Radiation Oncology, Biology, Physics, 82, 667–673. https://doi.org/10.1016/j.ijrobp.2010.10.071
Bai, J., Behera, M., & Bruner, D. (2017). A systematic review of the gut microbiome, treatment-related symptoms and targeted interventions in children with cancer. Supportive Care in Cancer, 26, 427–439. https://doi.org/10.1007/s00520-017-3982-3
Blankenberg, D., Gordon, A., Von Kuster, G., Coraor, N., Taylor, J., & Nekrutenko, A. (2010). Manipulation of FASTQ data with galaxy. Bioinformatics, 26, 1783–1785. https://doi.org/10.1093/bioinformatics/btq281
Blum, H.E. (2017). The human microbiome. Advances in Medical Sciences, 62, 414–420. https://doi.org/10.1016/j.advms.2017.04.005
Boskey, E.R., Cone, R.A., Whaley, K.J., & Moench, T.R. (2001). Origins of vaginal acidity: High D/L lactate ratio is consistent with bacteria being the primary source. Human Reproduction, 16, 1809–1813.
Bronfenbrenner, U. (1979). The ecology of human development. Cambridge, MA: Harvard University Press.
Bruner, D.W., Lanciano, R., Keegan, M., Corn, B., Martin, E., & Hanks, G.E. (1993). Vaginal stenosis and sexual function following intracavitary radiation for the treatment of cervical and endometrial carcinoma. International Journal of Radiation Oncology, Biology, Physics, 27, 825–830.
Callahan, B.J., McMurdie, P.J., & Holmes, S.P. (2017). Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME Journal, 11, 2639–2643. https://doi.org/10.1038/ismej.2017.119
Callahan, B.J., McMurdie, P.J., Rosen, M.J., Han, A.W., Johnson, A.J., & Holmes, S.P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods, 13, 581–583. https://doi.org/10.1038/nmeth.3869
Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F.D., Costello, E.K., . . . Knight, R. (2010). QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7, 335–336. https://doi.org/10.1038/nmeth.f.303
Caporaso, J.G., Lauber, C.L., Walters, W.A., Berg-Lyons, D., Huntley, J., Fierer, N., . . . Knight, R. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME Journal, 6, 1621–1624. https://doi.org/10.1038/ismej.2012.8
Caporaso, J.G., Lauber, C.L., Walters, W.A., Berg-Lyons, D., Lozupone, C.A., Turnbaugh, P.J., . . . Knight, R. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences, 108(Suppl. 1), 4516–4522. https://doi.org/10.1073/pnas.1000080107
Champer, M., Wong, A.M., Champer, J., Brito, I.L., Messer, P.W., Hou, J.Y., & Wright, J.D. (2018). The role of the vaginal microbiome in gynaecological cancer. BJOG, 125, 309–315. https://doi.org/10.1111/1471-0528.14631
Chase, D., Goulder, A., Zenhausern, F., Monk, B., & Herbst-Kralovetz, M. (2015). The vaginal and gastrointestinal microbiomes in gynecologic cancers: A review of applications in etiology, symptoms and treatment. Gynecologic Oncology, 138, 190–200. https://doi.org/10.1016/j.ygyno.2015.04.036
Claesson, M.J., Clooney, A.G., & O’Toole, P.W. (2017). A clinician’s guide to microbiome analysis. Nature Reviews Gastroenterology and Hepatology, 14, 585–595. https://doi.org/10.1038/nrgastro.2017.97
Cote, M.L., Ruterbusch, J.J., Olson, S.H., Lu, K., & Ali-Fehmi, R. (2015). The growing burden of endometrial cancer: A major racial disparity affecting black women. Cancer Epidemiology, Biomarkers and Prevention, 24, 1407–1415. https://doi.org/10.1158/1055-9965.EPI-15-0316
Fettweis, J.M., Brooks, J.P., Serrano, M.G., Sheth, N.U., Girerd, P.H., Edwards, D.J., . . . Buck, G.A. (2014). Differences in vaginal microbiome in African American women versus women of European ancestry. Microbiology, 160, 2272–2282. https://doi.org/10.1099/mic.0.081034-0
Fredricks, D.N. (2011). Molecular methods to describe the spectrum and dynamics of the vaginal microbiota. Anaerobe, 17(4), 191–195. https://doi.org/10.1016/j.anaerobe.2011.01.001
Gajer, P., Brotman, R.M., Bai, G., Sakamoto, J., Schutte, U.M., Zhong, X., . . . Ravel, J. (2012). Temporal dynamics of the human vaginal microbiota. Science Translational Medicine, 4(132), 132ra152. https://doi.org/10.1126/scitranslmed.3003605
Goodrich, J.K., Di Rienzi, S.C., Poole, A.C., Koren, O., Walters, W.A., Caporaso, J.G., . . . Ley, R.E. (2014). Conducting a microbiome study. Cell, 158, 250–262. https://doi.org/10.1016/j.cell.2014.06.037
Green, K.A., Zarek, S.M., & Catherino, W.H. (2015). Gynecologic health and disease in relation to the microbiome of the female reproductive tract. Fertility and Sterility, 104, 1351–1357. https://doi.org/10.1016/j.fertnstert.2015.10.010
Hamady, M., & Knight, R. (2009). Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Research, 19, 1141–1152. https://doi.org/10.1101/gr.085464.108
Human Microbiome Project Consortium. (2012). A framework for human microbiome research. Nature, 486, 215–221. https://doi.org/10.1038/nature11209
Hummelen, R., Macklaim, J.M., Bisanz, J.E., Hammond, J.A., McMillan, A., Vongsa, R., . . . Reid, G. (2011). Vaginal microbiome and epithelial gene array in post-menopausal women with moderate to severe dryness. PloS One, 6(11), e26602. https://doi.org/10.1371/journal.pone.0026602
Johanson, G.A., & Brooks, G.P. (2010). Initial scale development: Sample size for pilot studies. Educational and Psychological Measurement, 70, 394–400. https://doi.org/10.1177/0013164409355692
Julious, S.A. (2005). Sample size of 12 per group rule of thumb for a pilot study. Pharmaceutical Statistics, 4, 287–291. https://doi.org/10.1002/pst.185
Kelly, D.L., Lyon, D.E., Yoon, S.L., & Horgas, A.L. (2016). The microbiome and cancer: Implications for oncology nursing science. Cancer Nursing, 39(3), E56–E62. https://doi.org/10.1097/NCC.0000000000000286
Knight, R., & Buhler, B. (Eds.). (2015). Follow your gut: The enormous impact of tiny microbes. New York, NY: Simon and Schuster.
Kyrgiou, M., Mitra, A., & Moscicki, A.B. (2017). Does the vaginal microbiota play a role in the development of cervical cancer? Translational Research, 179, 168–182. https://doi.org/10.1016/j.trsl.2016.07.004
Lozupone, C., & Knight, R. (2005). UniFrac: A new phylogenetic method for comparing microbial communities. Applied and Environmental Microbiology, 71, 8228–8235. https://doi.org/10.1128/aem.71.12.8228-8235.2005
Mandal, S., Van Treuren, W., White, R.A., Eggesbø, M., Knight, R., & Peddada, S.D. (2015). Analysis of composition of microbiomes: A novel method for studying microbial composition. Microbial Ecology in Health and Disease, 26, 27663. https://doi.org/10.3402/mehd.v26.27663
McDonald, D., Hyde, E., Debelius, J.W., Morton, J.T., Gonzalez, A., Ackermann, G., . . . Knight, R. (2018). American gut: An open platform for citizen science microbiome research. mSystems, 3(3). https://doi.org/10.1128/mSystems.00031-18
McElroy, K.G., Chung, S.Y., & Regan, M. (2017). CE: Health and the human microbiome: A primer for nurses. American Journal of Nursing, 117(7), 24–30. https://doi.org/10.1097/01.naj.0000520917.73358.99
Morgan, X.C., & Huttenhower, C. (2012). Chapter 12: Human microbiome analysis. PLOS Computational Biology, 8(12), e1002808. https://doi.org/10.1371/journal.pcbi.1002808
Muhleisen, A.L., & Herbst-Kralovetz, M.M. (2016). Menopause and the vaginal microbiome. Maturitas, 91, 42–50. https://doi.org/10.1016/j.maturitas.2016.05.015
Muls, A., Andreyev, J., Lalondrelle, S., Taylor, A., Norton, C., & Hart, A. (2017). Systematic review: The impact of cancer treatment on the gut and vaginal microbiome in women with a gynecological malignancy. International Journal of Gynecological Cancer, 27, 1550–1559. https://doi.org/10.1097/igc.0000000000000999
Navas-Molina, J.A., Peralta-Sánchez, J.M., Gonzalez, A., McMurdie, P.J., Vázquez-Baeza, Y., Xu, Z., . . . Knight, R. (2013). Advancing our understanding of the human microbiome using QIIME. Methods in Enzymology, 531, 371–444. https://doi.org/10.1016/b978-0-12-407863-5.00019-8
Olsen, G.J., Overbeek, R., Larsen, N., Marsh, T.L., McCaughey, M.J., Maciukenas, M.A., . . . Woese, C.R. (1992). The ribosomal database project. Nucleic Acids Research, 20(Suppl.), 2199–2200. https://doi.org/10.1093/nar/20.suppl.2199
Onderdonk, A.B., Delaney, M.L., & Fichorova, R.N. (2016). The human microbiome during bacterial vaginosis. Clinical Microbiology Reviews, 29, 223–238. https://doi.org/10.1128/cmr.00075-15
Piyathilake, C.J., Ollberding, N.J., Kumar, R., Macaluso, M., Alvarez, R.D., & Morrow, C.D. (2016). Cervical microbiota associated with higher grade cervical intraepithelial neoplasia in women infected with high-risk human papillomaviruses. Cancer Prevention Research, 9, 357–366. https://doi.org/10.1158/1940-6207.CAPR-15-0350
Rajagopala, S.V., Vashee, S., Oldfield, L.M., Suzuki, Y., Venter, J.C., Telenti, A., & Nelson, K.E. (2017). The human microbiome and cancer. Cancer Prevention Research, 10, 226–234. https://doi.org/10.1158/1940-6207.CAPR-16-0249
Ravel, J., Gajer, P., Abdo, Z., Schneider, G.M., Koenig, S.S., McCulle, S.L., . . . Forney, L.J. (2011). Vaginal microbiome of reproductive-age women. Proceedings of the National Academy of Sciences of the United States of America, 108(Suppl 1.), 4680–4687. https://doi.org/10.1073/pnas.1002611107
Renaud, G., Stenzel, U., Maricic, T., Wiebe, V., & Kelso, J. (2015). deML: Robust demultiplexing of Illumina sequences using a likelihood-based approach. Bioinformatics, 31, 770–772. https://doi.org/10.1093/bioinformatics/btu719
Schloss, P.D., Westcott, S.L., Ryabin, T., Hall, J.R., Hartmann, M., Hollister, E.B., . . . Weber, C.F. (2009). Introducing Mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75, 7537–7541. https://doi.org/10.1128/aem.01541-09
Sender, R., Fuchs, S., & Milo, R. (2016). Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell, 164, 337–340. https://doi.org/10.1016/j.cell.2016.01.013
Sinha, R., Abnet, C.C., White, O., Knight, R., & Huttenhower, C. (2015). The microbiome quality control project: Baseline study design and future directions. Genome Biology, 16, 276. https://doi.org/10.1186/s13059-015-0841-8
Sinha, R., Abu-Ali, G., Vogtmann, E., Fodor, A.A., Ren, B., Amir, A., . . . Huttenhower, C. (2017). Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nature Biotechnology, 35, 1077–1086. https://doi.org/10.1038/nbt.3981
Spor, A., Koren, O., & Ley, R. (2011). Unravelling the effects of the environment and host genotype on the gut microbiome. Nature Reviews Microbiology, 9, 279–290. https://doi.org/10.1038/nrmicro2540
Tang, Z.Z., Chen, G., & Alekseyenko, A.V. (2016). PERMANOVA-S: Association test for microbial community composition that accommodates confounders and multiple distances. Bioinformatics, 32, 2618–2625. https://doi.org/10.1093/bioinformatics/btw311
Touchefeu, Y., Montassier, E., Nieman, K., Gastinne, T., Potel, G., Bruley des Varannes, S., . . . de La Cochetière, M.F. (2014). Systematic review: The role of the gut microbiota in chemotherapy- or radiation-induced gastrointestinal mucositis—Current evidence and potential clinical applications. Alimentary Pharmacology and Therapeutics, 40, 409–421. https://doi.org/10.1111/apt.12878
Ursell, L.K., Metcalf, J.L., Parfrey, L.W., & Knight, R. (2012). Defining the human microbiome. Nutrition Reviews, 70(Suppl. 1), S38–S44. https://doi.org/10.1111/j.1753-4887.2012.00493.x
van Belle, G. (2002). Statistical rules of thumb. New York, NY: John Wiley and Sons.
Vázquez-Baeza, Y., Pirrung, M., Gonzalez, A., & Knight, R. (2013). EMPeror: A tool for visualizing high-throughput microbial community data. GigaScience, 2, 16. https://doi.org/10.1186/2047-217X-2-16
Werner, J.J., Koren, O., Hugenholtz, P., DeSantis, T.Z., Walters, W.A., Caporaso, J.G., . . . Ley, R.E. (2012). Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. International Society for Microbial Ecology Journal, 6, 94–103. https://doi.org/10.1038/ismej.2011.82
Wilson, I.D., & Nicholson, J.K. (2017). Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Translational Research, 179, 204–222. https://doi.org/10.1016/j.trsl.2016.08.002
Zitvogel, L., Ayyoub, M., Routy, B., & Kroemer, G. (2016). Microbiome and anticancer immunosurveillance. Cell, 165, 276–287. https://doi.org/10.1016/j.cell.2016.03.001


