Enfortumab Vedotin: Nursing Perspectives on the Management of Adverse Events in Patients With Locally Advanced or Metastatic Urothelial Carcinoma
Background: Many patients with locally advanced or metastatic urothelial carcinoma (mUC) need additional treatment options beyond PD-1 or PD-L1 inhibitors and platinum-based chemotherapies. Enfortumab vedotin-ejfv (EV) is an antibody–drug conjugate directed at Nectin-4 that received accelerated approval for treatment of adults with locally advanced or mUC previously treated with PD-1/PD-L1 inhibitors and platinum- containing chemotherapy in the neoadjuvant/adjuvant, locally advanced, or metastatic settings.
Objectives: This article provides practical considerations and recommendations regarding common and potentially treatment-limiting adverse events that may arise with EV therapy.
Methods: The clinical data that supported the approval of EV are reviewed, and supporting safety and management considerations are provided based on the authors’ experience.
Findings: EV therapy can be optimized through patient and caregiver education, proactive patient monitoring, early identification of adverse events, and timely intervention to alleviate symptoms.
Jump to a section
Locally advanced or metastatic urothelial carcinoma (mUC) is an aggressive and incurable disease that disproportionately affects older adults, often those with a history of smoking and comorbidities, including cardiovascular disease and diabetes. Safe and effective treatment options are limited. Platinum-based chemotherapy, the standard initial therapy for mUC, is often difficult to tolerate and responses often are short-lived. In the second-line setting, approved programmed cell death protein-1 (PD-1) or programmed cell death-ligand 1 (PD-L1) inhibitor therapies provide meaningful responses in 13%–29% of patients with mUC (Balar et al., 2017; Bristol-Myers Squibb, 2020; EMD Serono, 2019; Merck, 2020; Powles et al., 2017; Rosenberg et al., 2016). Subsequent therapies, including single-agent taxanes, have low objective response rates of only 11%–13% (Bellmunt et al., 2017; Powles et al., 2018). Therefore, a great unmet need for effective treatment options exists throughout the mUC treatment journey.
Enfortumab vedotin-ejfv (EV) received accelerated approval from the U.S. Food and Drug Administration (FDA) in December 2019 for treatment of adults with locally advanced or mUC previously treated with a PD-1 or PD-L1 inhibitor and a platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced, or metastatic setting (Astellas Pharma, 2019). EV is an antibody–drug conjugate (ADC) that comprises monomethyl auristatin E (MMAE) as the active anticancer component (Challita-Eid et al., 2016; Doronina et al., 2003; Liu et al., 2020). MMAE induces cell death by disrupting the microtubule apparatus. Unlike other microtubule-disrupting agents, such as taxanes and vinca alkaloids, EV is designed to target the delivery of MMAE to specific cells by using an antibody-delivery mechanism directed against Nectin-4, which is highly expressed in UC and involved in cellular processes associated with oncogenesis (Challita-Eid et al., 2016; Doronina et al., 2003; Liu et al., 2020). EV is administered via IV on days 1, 8, and 15 of each 28-day cycle.
EV-201 (NCT03219333) is a global, phase 2, single-arm study of EV in patients with locally advanced or mUC previously treated with platinum-containing chemotherapy and anti–PD-1/PD-L1 therapy (cohort 1) or anti-PD-1/PD-L1 therapy in patients who are platinum-naive and cisplatin-ineligible (cohort 2) (Rosenberg et al., 2019). In cohort 1, the basis for the FDA’s accelerated approval of EV, the objective response rate was 44%, with 12% complete response per Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1. The median response duration was 7.6 months, and the estimated median progression-free survival and overall survival were 5.8 months and 11.7 months, respectively. With an additional year of follow-up, median overall survival was extended to 12.4 months (O’Donnell et al., 2020). In the confirmatory randomized phase 3 trial (EV-301), median overall survival with EV was 12.88 months, as compared to 8.97 months for patients randomized to standard chemotherapy (Powles et al., 2021).
As with many anticancer therapies, EV is associated with adverse events (AEs). The most common treatment-emergent AEs across the EV trials were peripheral neuropathy (56%), fatigue (56%), decreased appetite (52%), rash (52%), alopecia (50%), nausea (45%), diarrhea (42%), dysgeusia (42%), dry eyes (40%), dry skin (26%), pruritis (26%), and vomiting (18%) (Astellas Pharma, 2019). The 16% discontinuation rate related to AEs (Astellas Pharma, 2019), which is consistent with other therapies in this population (Bellmunt et al., 2017; Powles et al., 2018), suggests that these events were generally manageable. A case study for managing treatment-emergent AEs is provided in Figure 1. 
Clinical Management of Adverse Events
This article provides practical recommendations to help nurses, advanced practitioners, and other clinicians manage select EV-related AEs based on published literature and guidelines when available, as well as expert opinion and clinical experience. Always refer to the current EV prescribing information for the most up-to-date dose modification and toxicity management guidance as new data becomes available. Summaries of key nursing and patient education considerations are provided in Figures 2 and 3, respectively. Although thorough assessment is essential to identify and address any AE or functional decline, this article focuses on skin reactions, peripheral neuropathy, ocular disorders, and hyperglycemia, with additional discussion of gastrointestinal and hematologic toxicities. With proactive monitoring, identification, and management of these AEs, nurses can help prevent or alleviate symptoms that might otherwise lead to treatment interruption or discontinuation. 
Skin Reactions
Skin reactions are anticipated during EV treatment because of the presence of low-to-moderate levels of Nectin-4 in skin keratinocytes, sweat glands, and hair follicles (Challita-Eid et al., 2016). In the EV clinical trials, skin reactions occurred in 54% of EV-treated patients (26% maculopapular rash, 30% pruritus), including 10% with grade 3–4 reactions that included symmetrical drug-related intertriginous and flexural exanthema, bullous dermatitis, exfoliative dermatitis, and palmar-plantar erythrodysesthesia (Astellas Pharma, 2019). Treatment-related rash often presented during the first cycle in EV-201 cohort 1, with a median onset of 0.53 months (range = 0.03–7.39) from the start of treatment (Rosenberg et al., 2019). At the time of last follow-up, among those who experienced rash, 73% had complete resolution (median = 0.72 months, range = 0.03–2.66), 20% showed improvement (median = 0.72 months, range = 0.03–7.2), and those with ongoing rash had predominantly grade 1 (75%). 
Although maculopapular rash and pruritus were the most commonly reported skin reactions, presentation was variable in terms of type of reaction (i.e., rash, dry skin, pruritus, and hyper/hypopigmentation), type of rash (i.e., maculopapular, pustular, erythematous, and bullous), distribution (i.e., localized or widespread), location (i.e., chest, back, arms, thighs, axillae), and characterization (i.e., itchy and/or painful). Although skin reactions often occur early, they may occur or recur at any time during treatment. Most skin reactions are mild and transient; however, serious presentations (including mucosal involvement, bullous lesions, or exfoliation) require prompt referral to dermatology for management based on specific etiology and diagnosis.
Risk factors: Currently, there are no identified risk factors for developing skin reactions while on EV treatment. In general, characteristics that may predispose patients to skin reactions include prior history of a dermatologic condition, rash/pruritus, allergies, dry skin, immunosuppression, and/or high sun exposure.
Monitoring/Prevention: Advise patients of the potential for skin reactions, common presentations, and the importance of early reporting. For patients with a skin reaction, full-body examination helps ensure accurate estimation of the affected body surface area. Treating mild skin reactions allows for more rapid and effective management, potentially preventing development of severe skin reactions that could lead to treatment delays.
General advice for skin care during and after cancer treatment includes sunscreen (SPF 30 or greater and free of para-aminobenzoic acid); lukewarm rather than hot showers; proper hydration; frequent use of emollients; mild detergents and skin cleansers; alcohol-free, fragrance-free hypoallergenic moisturizers; hypoallergenic makeup; and avoidance of over-the-counter (OTC) acne medications (Bensadoun et al., 2013).
Interventions: As detailed in Table 1, dose interruption, reduction, and/or permanent discontinuation is recommended for grade 3–4 skin reactions, based on National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 4.03, and body surface area estimation. 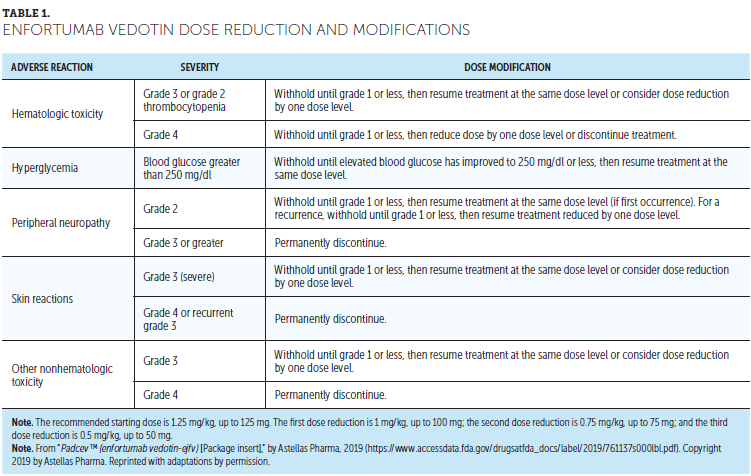
Pharmacologic therapies for rash include topical corticosteroids, such as OTC hydrocortisone (low potency), triamcinolone (medium potency), and clobetasol (high potency); oral or topical antihistamines (diphenhydramine or less-sedating antihistamines, such as cetirizine) (Wu & Adamson, 2019); and systemic corticosteroids for severe cases (Rosenberg et al., 2019; Salzmann et al., 2019; Wu & Adamson, 2019). Topical antibiotics or antifungals may be needed to treat secondary infections. Nonpharmacologic strategies for anticancer treatment-induced rash include fragrance-free moisturizers applied twice daily (ideally within 15 minutes after showering/bathing). In general, gentle, unscented creams and emollients, such as white petrolatum at least twice daily, are recommended prophylactically and after rash appears (Bensadoun et al., 2013). Creams containing anti-itch ingredients, such as pramoxine, camphor, menthol, or oatmeal, may be helpful for itchy rash (Pernambuco-Holsten, 2013).
A referral to dermatology for evaluation and further management is indicated for skin reactions that exceed 30% of body surface area (grade 3 or higher), involve the mucosa, bullous lesions, or exfoliation, or do not respond to a combination of steroids, antihistamines, and dose modifications. Early referral to dermatology for lower-grade skin reactions is also a reasonable approach for proactive evaluation and management.
Peripheral Neuropathy
Peripheral neuropathy (PN) is a clinically relevant adverse effect of several anticancer therapies, including platinum- and taxane-based chemotherapies and MMAE-containing ADC agents, that may limit treatment duration and impair quality of life (Hershman et al., 2014; Masters et al., 2018). In EV-201 cohort 1, treatment-related PN occurred in 50% of patients, with sensory PN (i.e., pain/burning, numbness, tingling, or loss of sensation) reported more frequently (44%) than motor PN (i.e., loss of coordination and/or muscle weakness) (14%) (Rosenberg et al., 2019). Most PN was grade 1–2, and onset occurred at a median of 2.43 months (range = 0.03–7.39) after starting treatment. At last evaluation, 76% of patients who experienced any grade PN had resolution or ongoing grade 1 PN, with a median of 1.18 months (range = 0.26–4.86) to improvement and 1.48 months (range = 0.23–11.6) to resolution.
Risk Factors: In addition to certain anticancer therapies, risk factors for PN include comorbidities (e.g., diabetes mellitus), older age, spinal involvement of mUC, and nonmalignant spinal disease. In EV-201 cohort 1, where grade 2 or higher PN was an exclusion criterion, 52% of patients with PN at baseline had worsening PN during treatment, similar to the rate of PN in the overall population (Rosenberg et al., 2019). Therefore, preexisting PN does not appear to be a risk factor for worsening PN with EV therapy.
Monitoring/Prevention: Early identification and management of PN is essential. Assess and document symptoms, perform a thorough neurologic and musculoskeletal evaluation, and assess impact on daily function at baseline and at each clinical visit. Table 2 provides a checklist for PN assessment, which can be helpful for evaluating the extent and severity of PN. Referral to specialty providers may be necessary to assess for other contributing factors, such as vascular evaluation of circulatory etiologies. 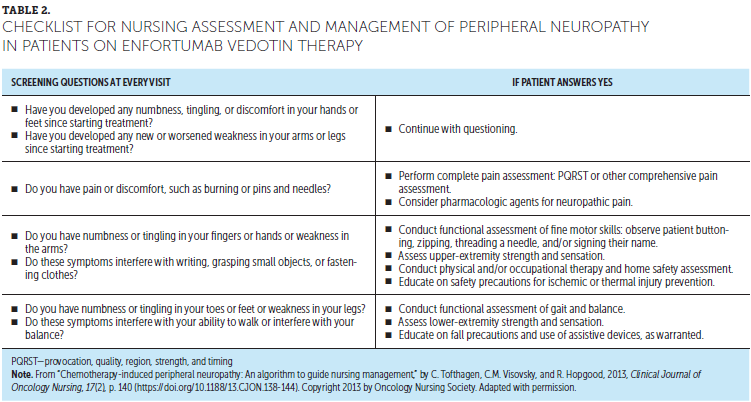
Patients have different levels of tolerance for PN symptoms, and underreporting the magnitude of PN is a concern among patients wishing to avoid treatment delay or discontinuation. In the authors’ experience, early detection and management has the potential to allow some recovery from symptoms and prevent worsening PN. Educate patients and caregivers about signs and symptoms of PN (sensory PN: pain/burning, numbness, tingling, or loss of sensation; motor PN: loss of coordination, muscle weakness) and the importance of prompt reporting and management to reduce risk of severe and potentially irreversible symptoms.
Interventions: EV-related PN is managed using a combination of dose interruptions, reductions, and discontinuation using CTCAE grading criteria. Dose interruption is recommended at the first sign of grade 2 PN until improvement to grade 1 or resolution. For a recurrent episode, EV should be held, then resumed at a lower dose. Grade 3 or higher PN warrants permanent discontinuation of EV. In addition, significant or refractory PN justifies referral to neurology for evaluation and management.
For patients with grade 2 or lower PN who continue on EV, consider adjunctive pharmacologic therapy for PN symptoms. Although there are no agents specifically recommended for managing EV-associated PN, the American Society of Clinical Oncology (ASCO) and joint European expert clinical practice guidelines recommend duloxetine and other alternatives for painful PN (Hershman et al., 2014; Jordan et al., 2020).
Nonpharmacologic therapies may be used for PN, but evidence to support their efficacy is lacking. Physical and occupational therapies and rehabilitation support may help improve functional deficits resulting from PN (National Institute of Neurological Disorders and Stroke [NINDS], 2018). For motor PN, mechanical aids such as hand or foot braces may reduce physical disability and pain, improve gait disturbances, and help prevent foot injuries (NINDS, 2018).
Ocular Disorders
Ocular events occurred in 46% of EV-treated patients (Astellas Pharma, 2019). The majority of these events were associated with dry eyes, including blurred vision and corneal events of keratitis and limbal stem cell deficiency. Dry eye symptoms occurred in 36% of patients and blurred vision in 14% of patients, with a median onset of 1.9 months (range = 0.3–6.2) from starting treatment.
Risk Factors: Older age is a risk factor for dry eyes (Schaumberg et al., 2009). Ocular manifestations, such as keratitis and corneal ulcerations, have been reported with other anticancer therapies, including ADC agents (Eaton et al., 2015; Harman, 2016), and were exclusion criteria for the EV-201 trial. Contact lens use increases the risk of developing keratitis; therefore, favoring eyeglasses while on treatment is prudent.
Monitoring/Prevention: Baseline and routine eye examinations are not required (Astellas Pharma, 2019) but may be considered for patients with known ocular disorders. Artificial tears are recommended for prophylaxis of dry eyes and may help with reflexive tearing. In the authors’ experience, patients reported that blurry vision, dry eyes, and reflexive tearing can have a significant impact on quality of life and disrupt activities such as driving, reading, and watching television.
Interventions: Dose modifications for symptomatic ocular disorders are guided by the EV prescribing information regarding other nonhematologic toxicities. These include dose interruption for grade 3 events and permanent discontinuation for grade 4 events.
In general, grade 3 or lower dry eyes can be managed with artificial tears or ophthalmic topical steroids (if indicated after ophthalmic evaluation). Consider referral to ophthalmology/optometry for dry, watery eyes; blurred vision; eye pain; or other ocular symptoms that persist or recur. Advise patients and caregivers to alert their eye care professional about their EV therapy.
Hyperglycemia
In clinical trials, diabetes has been reported in 20% of patients with mUC (Galsky et al., 2018; Niegisch et al., 2018). Hyperglycemia in patients on anticancer therapy may be associated with acute and serious clinical scenarios, such as infection; acid-base, fluid, and electrolyte disorders; and progression to ketoacidosis, in addition to the long-term cardiovascular, renal, and neuropathic risks. In EV-201 cohort 1, patients with a history of diabetes mellitus were included, but those with uncontrolled diabetes (i.e., hemoglobin A1C of 8% or greater, or 7% or greater with associated diabetes symptoms) were excluded. At baseline, 15% of patients had hyperglycemia. Treatment-related hyperglycemia was observed in 11% of patients overall, including 32% of those with and 8% of those without preexisting hyperglycemia, with a median onset of 0.58 months (range = 0.26–9.23) from starting treatment (Rosenberg et al., 2019). Median time to improvement of hyperglycemia was 0.89 months (range = 0.59–1.18), and median time to resolution was 1.12 months (range = 0.26–6.47).
Risk Factors: When evaluating hyperglycemia in a patient on anticancer therapy, consider potential risk factors, including but not limited to diabetes mellitus, illness/infection, and use of systemic steroids. Hyperglycemia occurred in the EV clinical trials, and grade 3 or greater events increased consistently in patients with higher body mass index and higher baseline hemoglobin A1C (Astellas Pharma, 2019).
Monitoring/Prevention: Assessing baseline hemoglobin A1C is prudent before starting EV, and routine monitoring of nonfasting blood glucose levels is recommended prior to each EV dose. If hyperglycemia is present, investigate all potential etiologies, including steroid use and infection (Davies et al., 2018). Consider home glucose monitoring if pre-dose blood glucose values are increasing.
Patients with a history of diabetes or hyperglycemia should continue to see their endocrinologist/primary care provider and inform them of their EV therapy. For these patients, consider optimizing blood glucose control prior to starting EV as their cancer status allows. Educating patients about the potential for hyperglycemia, the importance of recognizing and reporting symptoms, and the potential for serious complications is essential. Advise patients to watch for and report frequent urination, increased thirst, blurred vision, confusion, drowsiness, loss of appetite, fruity breath smell, nausea, vomiting, or stomach pain. Counseling on lifestyle modifications, such as a healthy diet low in simple carbohydrates, regular exercise, and weight loss (if indicated) is sensible.
Interventions: Patients who experience hyperglycemia should be treated according to the local standard of care, including use of oral and/or injectable antihyperglycemic medication and consideration of referral to endocrinology. For nonfasting blood glucose greater than 250 mg/dl, withhold EV, regardless of the cause, until blood glucose has improved to 250 mg/dl or lower and then resume at the same dose level (Astellas Pharma, 2019).
Gastrointestinal Adverse Events
Patients on anticancer therapy often experience gastrointestinal AEs, such as nausea, diarrhea, vomiting, appetite loss, and dysgeusia, which can lead to decline in nutritional and hydration status. Ensuring adequate nutrition and hydration is essential to avoid complications, such as renal dysfunction, functional decline, fluid–electrolyte imbalance, and fatigue. Dose modifications for gastrointestinal AEs are guided by the EV prescribing information regarding other nonhematologic toxicities. Patients may better tolerate dysgeusia symptoms if they are aware of the potential for taste alterations and reduced appetite (Murtaza et al., 2017). Supportive care strategies include using lemon juice and chewing gum prior to meals; having small frequent meals, good oral hygiene; drinking water with meals; using plastic instead of metal utensils; using more/less salt and flavoring for food; and avoiding foods with strong smells (Murtaza et al., 2017; Rehwaldt et al., 2009). Zinc supplements may be helpful for dysgeusia, but current evidence is conflicting (Lyckholm et al., 2012; Murtaza et al., 2017).
Hematologic Adverse Events
Hematologic toxicities may occur at any time during anticancer therapy. Dose interruption, reduction, and/or permanent discontinuation of EV is recommended for grade 3–4 hematologic laboratory abnormalities or grade 2 or greater thrombocytopenia. Although the incidence of treatment-related grade 3–4 hematologic toxicities in EV-201 cohort 1 was less than 10% (Rosenberg et al., 2019), these are important side effects requiring patient education and nurse management. Regular monitoring is necessary to detect hematologic laboratory abnormalities, including neutropenia, anemia, and thrombocytopenia.
Implications for Nursing
Oncology nurses, including bedside, infusion, clinical trials, advanced practice, and oncology nurse navigators, have an essential role in monitoring and managing AEs in patients undergoing cancer treatment, as well as educating patients and caregivers about expectations regarding AEs. Nursing assessment, including discussion with patients and caregivers, active and passive physical examination, and review of laboratory assessments, is critical for the safe care of patients receiving EV. Given the weekly administration schedule, nurses are positioned to play a critical role in identifying and managing treatment-emergent AEs. Through careful monitoring and prompt intervention, most EV-related AEs can be mitigated with medications, self-care, and/or dose modifications, potentially maximizing patient benefit from this promising therapy. 
Conclusion
EV monotherapy has consistently shown activity in patients with locally advanced UC or mUC, and therapeutic clinical trials are ongoing in other disease settings and in combination with other anticancer agents. Nursing familiarity with this drug, its effects, and appropriate patient monitoring and management is poised to become increasingly important in the clinical oncology setting.
About the Author(s)
Amanda Pace, BSN, RN, OCN®, is a research nurse in genitourinary medical oncology at the Dana-Farber Cancer Institute in Boston, MA; Blaine Brower, MSN, RN, FNP-BC, is a nurse practitioner in genitourinary medical oncology at University of North Carolina Healthcare in Chapel Hill; Dawn Conway, BSN, RN, OCN®, is a nurse navigator in genitourinary oncology at University of Chicago Medicine in Illinois; and Dayna Leis, BSN, RN, OCN®, is a senior clinical research nurse in genitourinary medical oncology at NYU Langone Health in New York. Laurie LaRusso, MS, ELS, from Chestnut Medical Communications, provided writing support during development of the manuscript, which was paid for by Astellas-Seagen Inc. Nancy N. Chang, PharmD, and Jennifer Lill, PharmD, of Seagen Inc. also provided critical review and revision of the manuscript. The authors take full responsibility for this content. Pace and Brower received honoraria for participation in an Astellas-Seagen Inc. Bladder Cancer Advisory Board meeting. Brower, Conway, Leis, and Pace received honoraria from Astellas-Seagen Inc. for expert panel participation. Conway also has received consulting fees from Astellas-Seagen Inc. Brower is on the speaker’s bureau for enfortumab vedotin. The article has been reviewed by independent peer reviewers to ensure that it is objective and free from bias. Mention of specific products and opinions related to those products do not indicate or imply endorsement by the Oncology Nursing Society. Pace can be reached at apace2@partners.org, with copy to CJONEditor@ons.org. (Submitted July 2020. Accepted October 21, 2020.)